Research in the Baram Lab is carried out within a vibrant Neuroscience community. The lab is an integral part of the Conte Center (Conte Center@uci) which includes both human and animal model research, the UCI EpiCenter and the Center for Neurobiology of Learning and Memory (CNLM). Graduate students typically belong to the Department of Anatomy/Neurobiology. The lab has productive collaborations with addiction groups (Mahler), In vivo MR imaging (Obenaus), circuit mapping (Xu), hippocampal circuit computational approaches (Lynch), signal transduction of memory (Gall) and epigenomics and single cell transcriptomics (Mortazavi).
Maturation and function of the pleasure/reward circuitry are impacted by early-life adversity/stress:
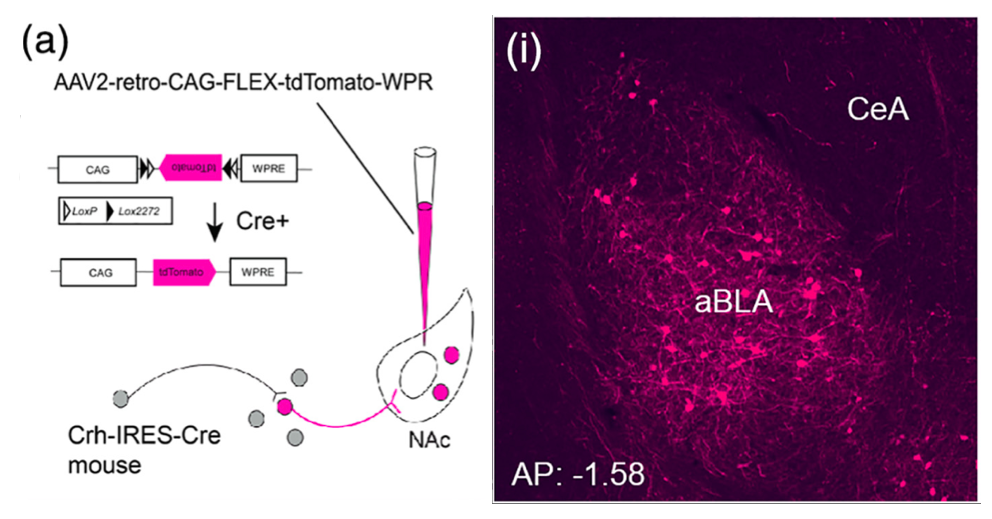
The reward circuitry underlies the fundamental processes of pleasure, reward, and happiness. Disruption of the function of this circuit contributes to serious mental health problems including drug addiction and depression. One potential manifestation of the aberrant function of the reward-related circuitry is anhedonia, the reduced ability to experience pleasure. Whereas anhedonia has traditionally been considered a component and predictor of depression, it has recently been validated as an independent biological dimensional entity that transects human schizophrenia, depression and the seeking of pro-hedonic opioid drugs. Remarkably, the molecular and circuit mechanisms of anhedonia and how it contributes to neuropsychiatric vulnerabilities are not fully understood. We have converging lines of evidence for alterations of the pleasure/reward circuitry as a result of early-life adversity. For example, we have discovered a novel, direct monosynaptic CRH+connection from basolateral amygdala (BLA) to the nucleus accumbens (NAc) that seem to be augmented after adversity (figures attached above; link to paper).
Ongoing experiments test the central hypotheses that(1) CRH+inputs from BLA to NAc strongly modulate NAc function; and (2) augmented activity of the CRH+BLA-NAc pathway after early-life adversity underlies the abnormal function of the reward circuit, presenting as anhedonia-like behaviors in adult mice.
We employ Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) and optogenetic technologies in CRH-IRES-CRE mice to test if NAc-directed CRH+inputs from BLA regulate pleasure/reward behaviors. We examine whether early-life adversity augments the CRH+ BLA-NAc connections, thus disrupting the function of the reward circuitry.
How early-life adversity modulates synapse development and persistence in stress-related brain circuits:
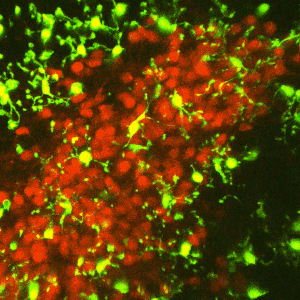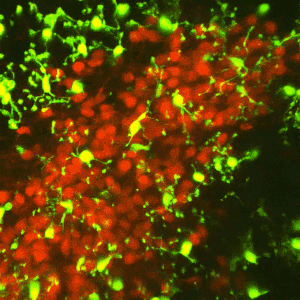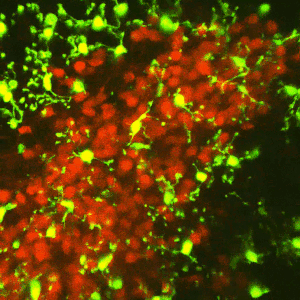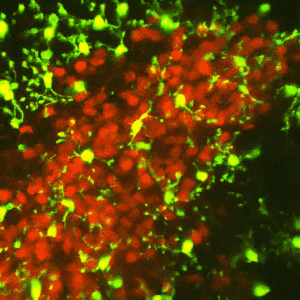
Early-life adversity can profoundly impact an individual’s risk for stress-related emotional disorders such as depression, likely by modulating the maturation of the underlying brain circuits. We find that early-life exposure to an impoverished environment provokes core symptoms of depression (anhedonia), accompanied by altered connectivity of stress-sensitive neurons. Specifically, we find an increase in the number of excitatory synapses onto corticotropin-releasing hormone (CRH)-expressing neurons in the paraventricular nucleus of the hypothalamus (PVN). Further, these synaptic changes suffice to induce enduring epigenomic changes in the expression of critical neuronal genes such as Crh. However, the mechanisms by which early-life adversity modulates synapse development and persistence in stress-related brain circuits remain unknown.Microglia, the brain’s resident immune cells, have emerged as key effectors in the shaping of synaptic connectivity in the developing visual and somatosensory systemsand hippocampus. Microglia are thus attractive candidates for playing a similar role in sculpting connectivity of stress-related hypothalamic neurons. In Dr. Jessica Bolton’s K99/R00 proposal, we aim to test this hypothesis during normal development, as well as during early-life adversity.
How early-life adversity/stress provokes spatial memory deficits and the transcriptional/epigenomic mechanisms that re-program hippocampal neurons and circuits:
Adverse early-life experiences (“stress”) are associated with lifelong cognitive deficits and risk of dementia, yet the underlying mechanisms remain unclear. The lab has designed and employed a potent model of early-life adversity, achieved by simulating poverty in rodent cages by limiting the amount of nesting and bedding in the cages. This model (LBN) has been adopted throughout the world. One week of rearing in the LBN cages leads to deficits in spatial memory that commence in adolescence and remain for life, progressively worsening with age.
Ongoing studies probe changes in gene expression in the hippocampus, the upstream regulatory mechanisms, and their contribution to memory deficits.
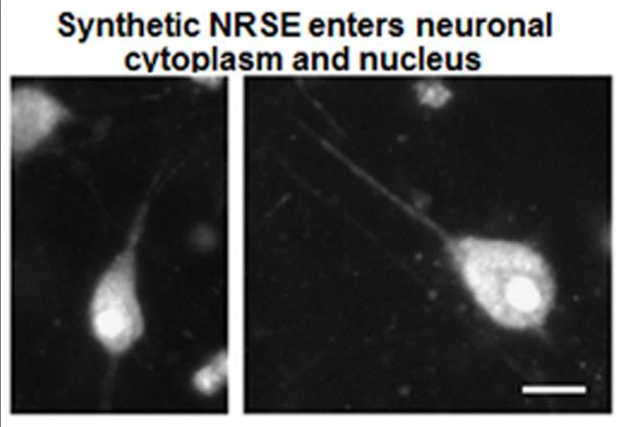
We have identified over a hundred differentially expressed (mostly downregulated) genes in the dorsal hippocampus. Transcription-factor target enrichment identified the stress-hormone receptor GR (glucocorticoid receptor) and, unexpectedly, the repressor neuron-restrictive silencer factor (NRSF/REST), as candidate upstream regulators. Blocking NRSF function transiently after the adversity period is being tested for rescuing hippocampus-dependent memory without influencing other behaviors (figures attached above; link to paper).
These studies are identifying a novel role for NRSF-mediated repression of crucial neuronal genes in early-life adversity-induced hippocampal dysfunction and enduring cognitive deficits. A better understanding of these mechanisms will enable the development of better therapeutics and preventative interventions for at-risk children.
How concurrent multiple acute stresses, such as occurring in mass shootings and natural disasters, impact memory processes in males and females:

Acute stress activates memory circuits to enhance memory and thus promote survival. Surprisingly, we recently discovered that multiple acute and concurrent stresses (MAS) impair spatial memory enduringly.
Concurrent acute concurrent physical, emotional and social stresses are typical of natural disasters and mass shootings. Therefore, uncovering the fundamental molecular, cellular and circuit mechanisms by which they impact memory networks is essential for elucidating the mental health impact of these events and identifying effective interventions.
We found that the severity of the MAS was not greater than that of a single stress that did not impair memory, at least when measured by plasma corticosterone (CORT) profiles and Fos activation of stress-sensitive hypothalamic neurons. However, MAS alone activated distinct brain circuits including amygdala-origin input pathways to the hippocampus. In addition, only MAS destroyed dendritic spines and synapses in dorsal hippocampal CA1, a subfield essential for spatial memory.
Probing these dramatic effects of MAS by sex, we discovered that females are vulnerable during high-estrogen cycle phases and protected when estrogen is low, suggesting a novel deleterious role for estrogen in females.
Ongoing studies test whether 1) the multiple hippocampal inputs engaged by MAS cause runaway activity in a reverberating CA3 network leading to the catastrophic collapse of hippocampal operations. 2) MAS unleash concerted activation of dendritic spine receptors for CORT, CRH, and estrogen on vulnerable spines, resulting in actin disruption and spine/synapse loss.
Computational, electrophysiological, functional/ behavioral, chemogenetic and optogenetic methods are used, together with unique transgenic mice lacking specifically the membrane or nuclear forms of estrogen receptors.
How prolonged early-life seizures, especially those associated with fever, disrupt the maturation of hippocampal circuits and memory processes:

Seizures are the most overt manifestation of epilepsy, the 3rd most common chronic brain disorder. However, epileptic seizures are commonly accompanied by problems in memory that contribute greatly to poor quality of life. Cognitive problems are especially troubling in temporal lobe epilepsy (TLE), where seizures involve hippocampal-cortical circuits. Although the seizures in TLE have been considered the cause of the memory problems, recent studies show that memory deficits often arise before, or independent of, the onset of spontaneous seizures.
TLE most commonly arises in individuals with a history of prolonged childhood febrile seizures (febrile status epilepticus, FSE), and FSE is increasingly recognized to directly provoke hippocampal injury and impair memory. Therefore, understanding the mechanisms of FSE-related cognitive problems is highly impactful.
We have generated a rat model of experimental (e)FSE that provokes both TLE-like epilepsy as well as memory deficits, thus providing a potent tool to probe these issues. Recently, we have identified at the molecular, cellular and circuit levels some of the mechanisms underlying eFSE-induced memory problems. In addition to inflammatory changes, we found that eFSE provokes coordinated transcriptionally-regulated changes in the expression of a relatively small set of genes that govern neuronal behavior. These changes result from augmented function of the neuron-restrictive silencing factor (NRSF) after eFSE. This transcription factor has unique roles in neuronal maturation and in the function of mature neurons. Accordingly, blocking NRSF chromatin-binding for a short period after eFSE model abrogated the cognitive deficits, positioning NRSF as an exciting potential target for translational interventions (figures attached above; link to paper).
Ongoing studies probe the mechanisms of the rescue of cognitive functions by NRSF blockade in the developing hippocampal-cortical circuit.
Whereas ~500 hippocampal genes are potential targets of NRSF, increased NRSF levels after eFSE repressed only ~30 in the hippocampus, governed by the affinity of these genes to NRSF. Focusing on the developing dentate gyrus granule cells (DG-GCs), we test the key role of NRSF in eFSE-mediated structural and functional defects, as will be uncovered by obtaining RNA-seq at single-cell resolution as well as NRSF-ChIP seq.
Intra-individual methylomics in rodents and humans as predictive biomarkers of early-life adversity-induced cognitive and emotional problems:
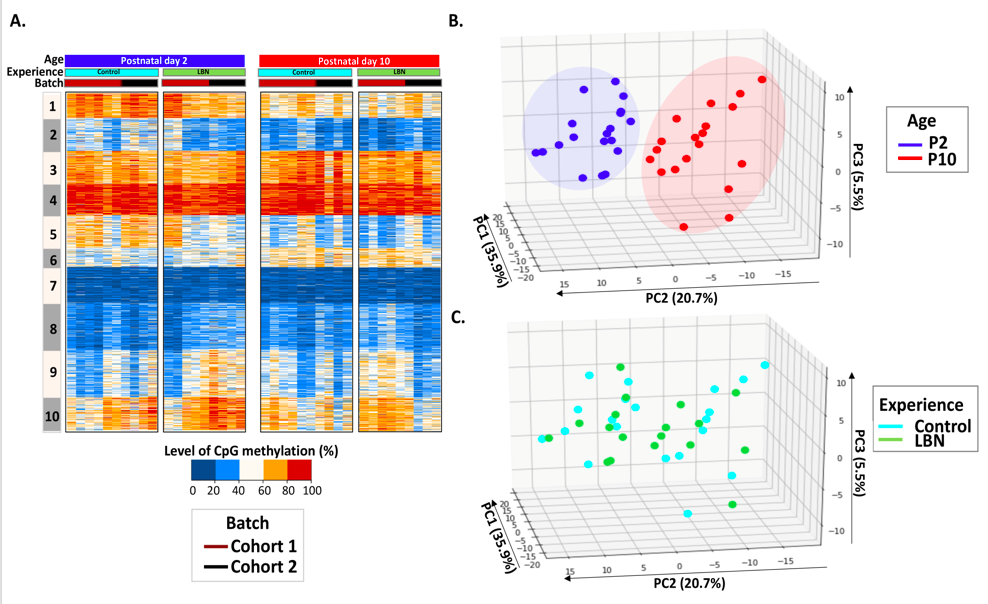
Genetic and environmental factors interact during sensitive periods early in life to influence mental health and disease via epigenetic processes such as DNA methylation. However, it is not known if DNA methylation changes outside the brain provide an ‘epigenetic signature’ of early-life experiences. In addition, large inter-individual variability often hampers the detection of experience-dependent and predictive changes of the methylome.
The lab employs a novel intra-individual approach by measuring DNA methylation from buccal cells of individual rats before and immediately after exposure to one week of typical or adverse early-life experience. We find that whereas inter-individual changes in DNA methylation reflect the effect of age, DNA methylation changes within paired DNA samples from the same individual reflect the impact of diverse neonatal experiences (figures attached above; link to paper).
Ongoing studies are now examining intra-individual methylation profiles in human babies sampled in the first weeks of life and again at 1 year of age. We know their intervening history and early-life environment, and are assessing the cognitive and emotional outcomes. Working with the Mortazavi lab and with computational and AI approaches, we hope to establish an intra-individual methylome signature as a potential predictive marker of outcome following early-life adversity.