
The Alzheimer’s disease (AD) genetic landscape identified microglia as a key disease-modifying cell type. Paired immunoglobulin-like type 2 receptor alpha (PILRA) is an immunoreceptor tyrosine-based inhibitory motif domain-containing inhibitory receptor, expressed by myeloid cells such as microglia. The known protective PILRA G78R gene variant reduces AD risk in apolipoprotein E4 (APOE4) carriers and is enriched in a cohort of healthy centenarians. However, mechanisms underlying protective effects in microglia are undefined. In this study, co-led by researchers from Denali Therapeutics and Blurton-Jones Lab, we identified biological functions of PILRA in human induced pluripotent stem cell-derived microglia (iMG) and chimeric AD mice. PILRA knockout (KO) in iMG rescued ApoE4-mediated immunometabolic deficits and prevented lipotoxicity through increased lipid storage, improved mitochondrial bioenergetics, and antioxidant activity. PILRA KO also enhanced microglial chemotaxis and attenuated inflammation. With pharmacological inhibitor studies, we showed that peroxisome proliferator-activated receptor and signal transducer and activator of transcription 1/3 mediated PILRA-dependent microglial functions. AD mice transplanted with human PILRA KO microglia exhibited reduced amyloid pathology and rescued synaptic markers. A high-affinity ligand blocking PILRA antibody phenocopied PILRA KO iMG. These findings suggest that PILRA is a pharmacologically tractable therapeutic target for AD.





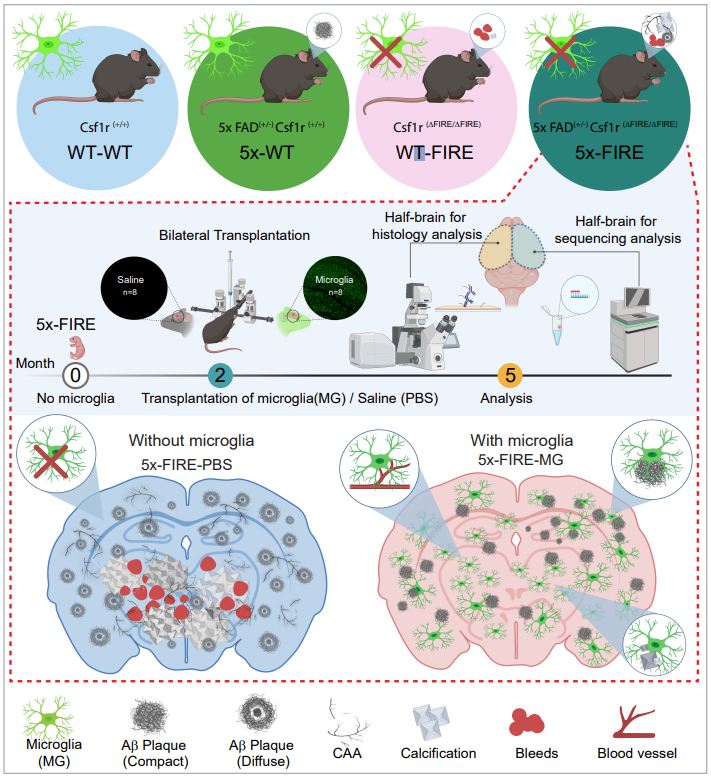

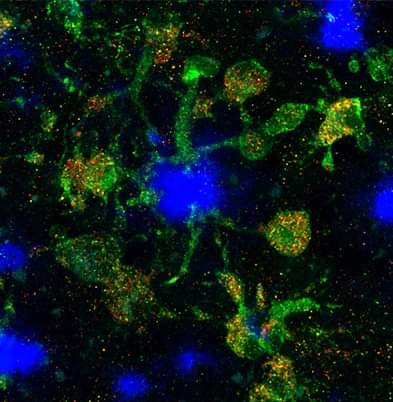 Background: Disease-associated microglia (DAMs), that surround beta-amyloid plaques, represent a transcriptionally-distinct microglial profile in Alzheimer’s disease (AD). Activation of DAMs is dependent on triggering receptor expressed on myeloid cells 2 (TREM2) in mouse models and the AD TREM2-R47H risk variant reduces microglial activation and plaque association in human carriers. Interestingly, TREM2 has also been identified as a microglial lipid-sensor, and recent data indicates lipid droplet accumulation in aged microglia, that is in turn associated with a dysfunctional proinflammatory phenotype. However, whether lipid droplets (LDs) are present in human microglia in AD and how the R47H mutation affects this remains unknown.
Background: Disease-associated microglia (DAMs), that surround beta-amyloid plaques, represent a transcriptionally-distinct microglial profile in Alzheimer’s disease (AD). Activation of DAMs is dependent on triggering receptor expressed on myeloid cells 2 (TREM2) in mouse models and the AD TREM2-R47H risk variant reduces microglial activation and plaque association in human carriers. Interestingly, TREM2 has also been identified as a microglial lipid-sensor, and recent data indicates lipid droplet accumulation in aged microglia, that is in turn associated with a dysfunctional proinflammatory phenotype. However, whether lipid droplets (LDs) are present in human microglia in AD and how the R47H mutation affects this remains unknown.