Congratulations to Dr. Jaclyn Beck on her successful PhD dissertation defense “Innate and Adaptive Immunity in Aging and Alzheimer’s Disease.”
Shabestari et al publish in Cell Reports
Absence of microglia promotes diverse pathologies and early lethality in Alzheimer’s disease mice
Microglia are strongly implicated in the development and progression of Alzheimer’s disease (AD), yet their impact on pathology and lifespan remains unclear. Here we utilize a CSF1R hypomorphic mouse to generate a model of AD that genetically lacks microglia. The resulting microglial-deficient mice exhibit a profound shift from parenchymal amyloid plaques to cerebral amyloid angiopathy (CAA), which is accompanied by numerous transcriptional changes, greatly increased brain calcification and hemorrhages, and premature lethality. Remarkably, a single injection of wild-type microglia into adult mice repopulates the microglial niche and prevents each of these pathological changes. Taken together, these results indicate the protective functions of microglia in reducing CAA, blood-brain barrier dysfunction, and brain calcification. To further understand the clinical implications of these findings, human AD tissue and iPSC-microglia were examined, providing evidence that microglia phagocytose calcium crystals, and this process is impaired by loss of the AD risk gene, TREM2.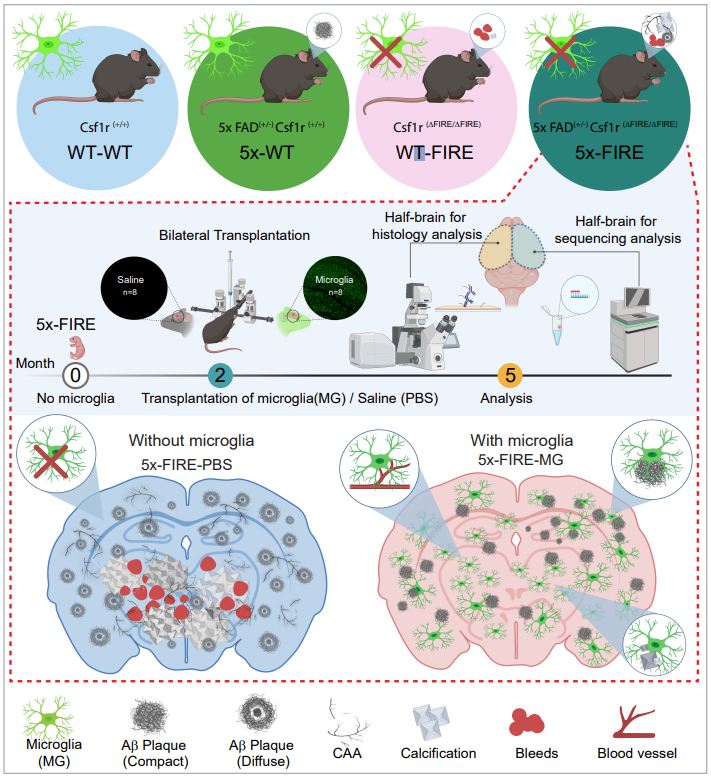
MBJ Lab at Keystone Symposia 2022
Senior scientist Dr. Hayk Davtyan, third-year graduate student Zahara Keulen, and second-year graduate student Jean Paul Chadarevian attend Keystone Symposia – Neuro-Immune Interactions in the Central Nervous System 2022!
Zahara presented a poster and talk titled “Neuronal Tau Pathology Alters Human Microglial Morphology, Transcriptome, and Function.” Jean Paul presented a poster and talk titled “Harnessing iPSC-Microglia to Deliver Therapeutics in the Brain.” MBJ-lab alumni Dr. Amanda Mcquade presented a poster and talk titled “Dampening Purinergic Signaling in TREM2-knockout iPSC-microglia Rescues Chemotactic Deficit.” Congratulations to everyone on their hard work and great feedback!









Congratulations Sepideh on your Advancement to Candidacy
On June 2, 2022, Sepideh Kiani Shabestari advanced to candidacy in the Blurton-Jones Lab with her PhD work investigating the relationship of Microglia and Amyloid Pathology in mice models. Congratulations!
Congratulations and Farewell to our PostDoc Dr. Alberto Granzotto!
Congratulations and farewell to our PostDoc Dr. Alberto Granzotto awarded the prestigious Best-Italian-Male-Post-Doc-In-The-MBJ-Lab-During-A-Pandemic two years in a row! We wish you continued success in your research.



Jairaman and McQuade et al publish in eLife
TREM2 regulates purinergic receptor-mediated calcium signaling and motility in human iPSC-derived microglia
The membrane protein TREM2 (Triggering Receptor Expressed on Myeloid cells 2) regulates key microglial functions including phagocytosis and chemotaxis. Loss-of-function variants of TREM2 are associated with increased risk of Alzheimer’s disease (AD). Because abnormalities in Ca2+ signaling have been observed in several AD models, we investigated TREM2 regulation of Ca2+ signaling in human induced pluripotent stem cell-derived microglia (iPSC-microglia) with genetic deletion of TREM2. We found that iPSC-microglia lacking TREM2 (TREM2 KO) show exaggerated Ca2+ signals in response to purinergic agonists, such as ADP, that shape microglial injury responses. This ADP hypersensitivity, driven by increased expression of P2Y12 and P2Y13 receptors, results in greater release of Ca2+ from the endoplasmic reticulum (ER) stores, which triggers sustained Ca2+ influx through Orai channels and alters cell motility in TREM2 KO microglia. Using iPSC-microglia expressing the genetically encoded Ca2+ probe, Salsa6f, we found that cytosolic Ca2+ tunes motility to a greater extent in TREM2 KO microglia. Despite showing greater overall displacement, TREM2 KO microglia exhibit reduced directional chemotaxis along ADP gradients. Accordingly, the chemotactic defect in TREM2 KO microglia was rescued by reducing cytosolic Ca2+ using a P2Y12 receptor antagonist. Our results show that loss of TREM2 confers a defect in microglial Ca2+ response to purinergic signals, suggesting a window of Ca2+ signaling for optimal microglial motility.
Behind a good mutation: How a gene variant protects against Alzheimer’s

Irvine, Calif., Feb. 14, 2022 — While the word “mutation” may conjure up alarming notions, a mutation in brain immune cells serves a positive role in protecting people against Alzheimer’s disease. Now University of California, Irvine biologists have discovered the mechanisms behind this crucial process. Their paper appears in the journal Alzheimer’s and Dementia.
The investigation centered on a variant of the PLCG2 gene, which makes the instructions for producing an enzyme important to brain immune cells called microglia. “Recently the mutation, which is known as P522R, was shown to lower the risk of developing late-onset Alzheimer’s,” said Hayk Davtyan, Ph.D., senior researcher in the laboratory of Mathew Blurton-Jones, professor of neurobiology & behavior, where the study was conducted. The project was led by assistant project scientist Christel Claes, Ph.D., the paper’s first author.
The scientists used CRISPR gene-editing technology to generate the protective mutation in human stem cells and then implanted microglia derived from those stem cells into humanized rodent models of Alzheimer’s disease.
“Our research showed for the first time that the P522R variant increased expression levels of several microglial genes that are reduced in people with Alzheimer’s. This provides some of the first evidence to explain how this protective mutation might reduce Alzheimer’s risk,” Davtyan said.
The variant also increased the number of T-cells, or white blood immune system cells, in the brain suggesting that it may increase the activation of other important aspects of immune function.
The results will help in designing further studies to understand exactly how microglia and T-cells interact to slow Alzheimer’s progression.
“Beyond that, the next step could be to identify drugs that can safely increase the activity of the PLCG2 enzyme and further promote protective microglial functions,” he said.
First author Christel Claes wonders whether a TREM2 stimulating antibody, like the one currently in a Phase 2 clinical study from Alector (AL002), could exert similar protection in AD patients as the P522R variant.
“It is well known that the PLCG2 P522R mutation increases TREM2 downstream signaling, an AD risk variant, thus it will be very interesting to study the effect of TREM2 stimulating antibodies on microglia-T cell crosstalk. Studies like ours pave the way to find new strategies to treat or prevent this disease that is taking such a toll on humanity, this is what drives us as neuroscientists.” said Davtyan.
The project was supported by a BrightFocus Postdoctoral Fellowship, the Cure Alzheimer’s Fund, National Institutes of Health and National Institute on Aging.
About the University of California, Irvine: Founded in 1965, UCI is the youngest member of the prestigious Association of American Universities and is ranked among the nation’s top 10 public universities by U.S. News & World Report. The campus has produced five Nobel laureates and is known for its academic achievement, premier research, innovation and anteater mascot. Led by Chancellor Howard Gillman, UCI has more than 36,000 students and offers 224 degree programs. It’s located in one of the world’s safest and most economically vibrant communities and is Orange County’s largest employer, contributing $7 billion annually to the local economy and $8 billion statewide. For more on UCI, visit www.uci.edu.
Media access: Radio programs/stations may, for a fee, use an on-campus ISDN line to interview UCI faculty and experts, subject to availability and university approval. For more UCI news, visit news.uci.edu. Additional resources for journalists may be found at communications.uci.edu/for-journalists.
Dr. Claes et al publish in Alzheimer’s and Dementia

The P522R variant of PLCG2, expressed by microglia, is associated with a reduced risk of Alzheimer’s disease (AD). Yet, the impact of this protective mutation on microglial responses to AD pathology remains unknown. Chimeric AD and wild-type mice were generated by transplanting PLCG2-P522R or isogenic wild-type human induced pluripotent stem cell microglia. At 7 months of age, single-cell and bulk RNA sequencing, and histological analyses were performed. The PLCG2-P522R variant induced a significant increase in microglial human leukocyte antigen (HLA) expression and the induction of antigen presentation, chemokine signaling, and T cell proliferation pathways. Examination of immune-intact AD mice further demonstrated that the PLCG2-P522R variant promotes the recruitment of CD8+ T cells to the brain. These data provide the first evidence that the PLCG2-P522R variant increases the capacity of microglia to recruit T cells and present antigens, promoting a microglial transcriptional state that has recently been shown to be reduced in AD patient brains.
Morgan Coburn, PhD Dissertation Defense
 Congratulations to Morgan Coburn on a successful Ph.D. Dissertation Defense “Using Chimeric Models to Study the Interactions between Human Microglia and Alzheimer’s Disease Pathology in vivo.“
Congratulations to Morgan Coburn on a successful Ph.D. Dissertation Defense “Using Chimeric Models to Study the Interactions between Human Microglia and Alzheimer’s Disease Pathology in vivo.“
Amanda McQuade Ph.D. Dissertation Defense
Congratulations to Amanda McQuade on a successful Ph.D. Dissertation Defense Development and application of a human microglia model to examine the influence of genetic risk factors on microglial function in Alzheimer’s disease:
Alzheimer’s disease (AD) is a progressive neurodegenerative disease for which there is no cure. Worldwide, AD is estimated to affect 50 million people with 10 million new cases each year. Developing therapeutics for Alzheimer’s disease has been particularly difficult given the complex etiology of this disease. Alzheimer’s disease is characterized by an accumulation of parenchymal beta-amyloid protein plaques, tau neurofibrillary tangles, and neuroinflammation. For sporadic AD, which accounts for around 95 % of AD cases, the direct trigger of neurodegeneration remains unclear. Understanding the mechanistic pathophysiology of this disease will allow for the development of more effective targeted therapeutics and biomarker studies to help patients.
In the last decade, genome-wide association studies (GWAS) have renewed interest in neuroinflammation as a potential disease-modifying mechanism. These large-scale genetic studies have uncovered a striking enrichment of immune-specific genes as risk-factors for Alzheimer’s disease. However, the function of many of these GWAS loci are not well understood. Additionally, many of these genes have poor homology between human cells and traditional murine disease-models. Thus, there is a critical need to develop a model of human microglia in order to understand how these immune risk factors influence human microglial function.
The focus of this dissertation is to develop and characterize a model of human microglia that can be readily studied without the need for isolation of human brain tissue surgically or post-mortem. Building on previous research in the lab, we have developed a model of human microglia differentiated from induced pluripotent stem cells (iPS-Microglia). This model follows developmental ontogeny with a primary differentiation into CD43+ hematopoietic progenitor cells before transition into a microglial differentiation medium containing neuron and astrocyte
derived cytokines to educate our microglia in a homeostatic brain-like environment. The result is a highly pure population of iPS-Microglia which perform key microglial functions and cluster alongside human microglial transcriptomes (Chapter 1).
Because this microglial model is fully defined beginning from iPS cells, it is possible to combine this approach with modern molecular biological manipulation such as CRISPR gene editing. This dissertation focuses on studies surrounding the AD-risk loci Triggering Receptor Expressed on Myeloid Cells II (TREM2). Predicted loss of function mutations in TREM2 increase Alzheimer’s disease risk up to 2-3 fold making it the highest microglial-specific risk factor for AD. By performing CRISPR-mediated knockout of TREM2 in human iPSCs and differentiating into
microglia, we find that TREM2-knockout locks microglia in a homeostatic state. Specifically, microglia lacking TREM2 are deficient in SYK-mediated phagocytosis, CXCR4-mediated migration, and are more sensitive to MCSF-mediated cell death.
In vivo, this translates to an inability to perform chemotaxis towards and compaction of beta-amyloid plaques leading to a build-up of pathology in the brain. We also characterize a distinct lack of transcriptional activation in TREM2 knockout cells suggesting that induction of the Disease Associated Microglia (DAM) profile may be an essential immune response in shielding the brain from dementia (Chapter 2).
Because TREM2-knockout cells are locked in a homeostatic state, they express high levels of homeostatic microglia markers P2RY12 and P2RY13. These purinergic receptors are critical for microglial communication with neurons and clearance of dead cells. We show that purinergic signaling has a profound effect on microglial motility and process extension and that hyper-expression and response of purinergic signaling in TREM2-knockout microglia may render these cells unable to sense gradients and activate against disease pathology. Furthermore, we highlight that partial inhibition of purinergic receptors will rescue migratory deficits characterized in TREM2-knockout microglia suggesting a therapeutic mechanism (Chapter 3).
Taken as a whole, the development of the iPS-Microglia model has yielded critical insight into the interaction of microglial function and the development of Alzheimer’s disease. This model has been readily adopted by many academic labs as well as pharmaceutical companies with the aim of studying human microglial function or other disease risk loci. By further studying AD risk genes in this fashion, we may be able to converge on several mechanisms by which microglial functions are able to attenuate or drive neurodegeneration. Focusing therapeutic efforts on these pathways could lead to the development of immunotherapies for Alzheimer’s disease.