The Thompson laboratory has been actively engaged in investigating the fundamental molecular and cellular events that underlie how the mutant Huntington’s disease gene causes degeneration of specific brain cell populations to induce motor and cognitive decline and premature death of patients with the ultimate goal to develop new therapeutic approaches. HD was one of the first neurodegenerative diseases for which a genetic cause was determined, and has served as a paradigm for researchers to study other such diseases. HD is an autosomal dominant neurodegenerative disease characterized by specific regions of neuronal dysfunction and loss, most notably of neurons in the striatum and cortex. The primary cause of HD is the expansion of a CAG triplet repeat encoding a polyglutamine (polyQ) tract within the amino terminal portion of the Huntingtin (Htt) protein. The mutation disrupts many cellular processes, including transcriptional regulation, vesicular trafficking, mitochondrial function, autophagic clearance, and protein modification, among others. The laboratory also focuses on understanding casual mechanisms that underlie Amyotrophic Lateral Sclerosis and more recently, X-linked Dystonia-Parkinsonism with the goal of developing treatment options for the disease. The research benefits from the integrated use of patient iPSCs and mouse models of disease together with the studies of RNA biology and network-based bioinformatics.
From structure to therapy: the TRiC Chaperonin network in Huntington’s disease
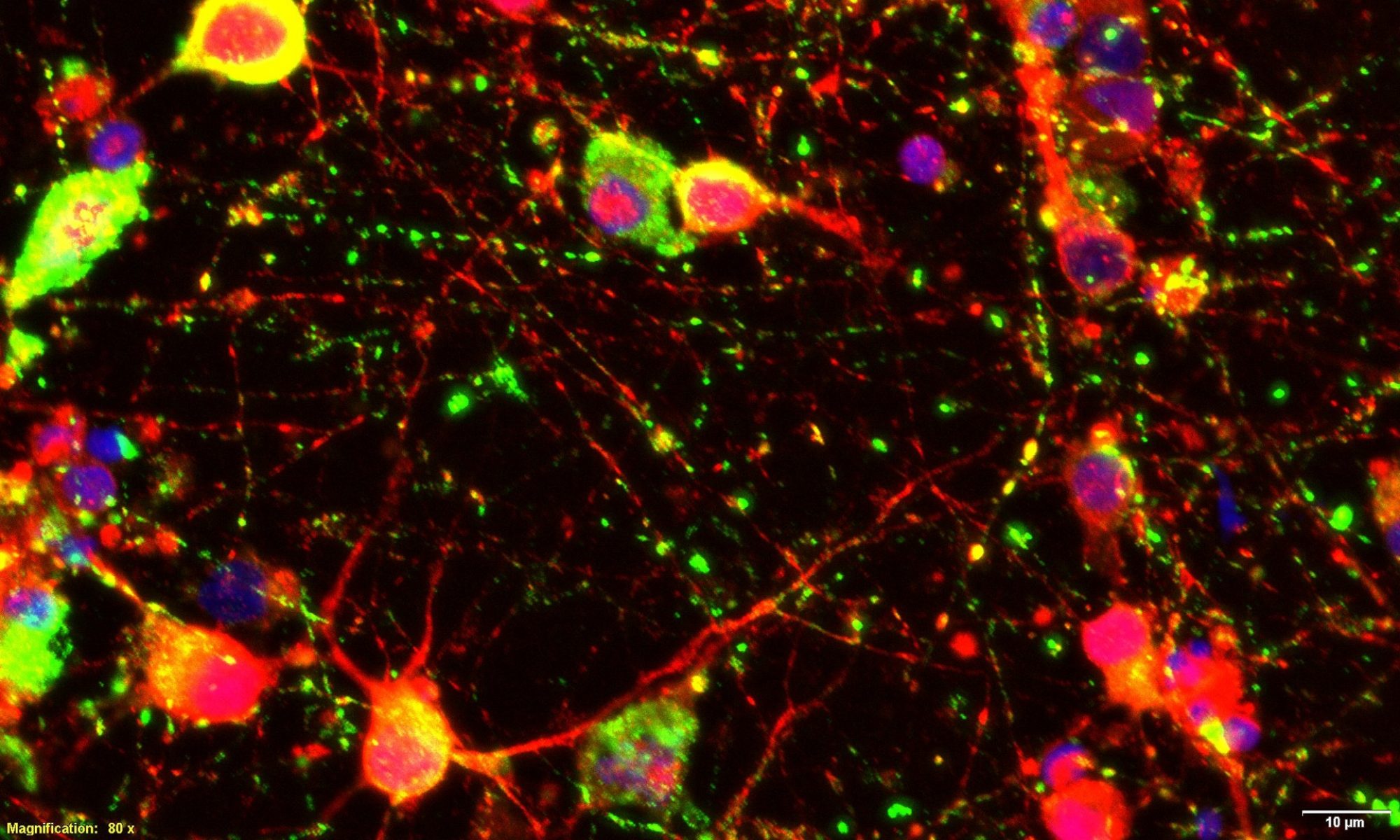
Exogenous ApiCCT1r enters cells and localizes to the cytosol and nucleus of PC12 cells.
The Thompson lab is a member of the Center for Protein Misfolding Consortium led by Dr. Wah Chui to investigate mechanisms of protein misfolding in neurodegenerative diseases, including HD, with an emphasis on translational medicine. Our work has shown that exogenous delivery of ApiCCT1 is sufficient to modulate HD phenotypes in vitro (Sontag, PNAS 2013). These data together with preliminary in vivo data suggest novel translational approaches to use TRiC reagents for HD therapeutics. Project 1: Mechanistic and structural basis for chaperonin-mediated remodeling of mutant HTT. Project leader, Wah Chiu (Baylor); Co-leader: Judith Frydman (Stanford). Understand, at a biochemical level, the interactions between toxic forms of the Htt protein and the TRiC network and use this understanding to suggest chaperone based strategies for intervention in HD. Project 1 investigates the structural and biochemical characteristics of mHTT species, in the absence and presence of TRiC reagents ex vivo, and in vitro and in vivo in HD cells and neurons. Project 2:The impact of TRiC reagents on mHTT-induced neuronal degeneration in vitro. Project leader: William Mobley (UCSD); Co-leader, Leslie M. Thompson (UCI). Understand the interactions between toxic forms of the Htt protein and the TRiC chaperonin system in cultured mammalian cells including neurons and from this understanding evaluate and optimize chaperone based interventions for HD. Project 2 investigates mHTT expression in creating deficits in neurons (BDNF trafficking, synapse function, gene expression, mitochondrial function, calcium homeostasis, proteostasis) and the effects of TRiC reagents in preventing/reducing these deficits. Project 3: TRiC modulation of CNS pathogenesis in HD mouse models. Project leader: Leslie Thompson (UCI), Co-leader, David Housman,(MIT). Project 3 will employ TRiC reagents in vivo to mitigate the impact of mHTT expression in mouse models of HD. Studies of mHTT species in the brains of HD model rodents will be investigated as will key elements of HD pathogenesis, including changes in BDNF trafficking and in gene expression. Researchers (Thompson Lab): J Overman and A Lau.
Epigenetic pathology and therapy in Huntington’s disease (Parent Institution – MIT: E. Fraenkel and D. Housman)
Our group was instrumental in describing interactions of the Huntingtin (HTT) protein with transcriptional regulatory proteins. We also showed that the interaction of mutant HTT with CREB-binding protein and other acetyltransferases could reduce their activity and that approaches to compensate for that decrease using “transcription-based therapies”, in this case HDAC inhibitors, were a rational approach to development of HD treatments. In collaboration with the Fraenkel and Housman labs, we have continued to study the underlying mechanisms of transcriptional dysregulation in HD. The first aim of this work is to establish baseline genome wide analyses of chromatin structure marks and transcription. We will extend RNAseq and ChIPseq analysis to a full-length mHTT mouse model and carry out relevant informatic analyses to further investigate the relationship between chromatin remodeling and transcriptional dysregulation in HD. Use translating ribosome affinity purification (TRAP) methodology to assess changes in chromatin structure and transcriptional from specific brain cell types in HD mouse models and controls. The second aim of this work is to evaluate targets for potential therapeutic intervention through modulation of the pathological epigenetic program in HD . Modulate the activity of targets from Aim 1 in differentiated HD iPS cells and in HD mouse models to assess their impact on epigenetic ensembles. Critically evaluate the hypothesis that the impact of mutant Htt on transcriptional dysregulation is the direct consequence of binding of mutant Htt to specific sites in chromatin. and modulate selected targets in HD mouse models to assess impact on organism health and pathology. Researchers (Thompson Lab): J. Stocksdale, L. Salazar, S. Winokur and R. Lim.
Neuron and Glial cellular signatures from normal and diseased iPS cells Library of Integrated Network-Based Cellular Signatures (LINCS):
Perturbation-induced Data and Signature Generation Centers (U54) S. Finkbeiner (Gladstone), C. Svendsen, J. Van Eyk, D. Sareen (Cedars), E. Fraenkel (MIT), J. Rothstein (JHU)
Induced Pluripotent Stem Cells marked with Nestin and Pax6
Major efforts have been initiated in the past five years to use induced pluripotent stem cells (iPSCs) derived from HD and control subjects to study HD, and the Thompson lab is part of a consortium of investigators to use these iPSCs as a platform for drug discovery and mechanistic studies. Initial studies show that a number of key molecular alterations are present in the differentiated iPS cells, including transcriptional dysregulation analyzed at the genomic level. We recently uncovered a novel signature in mouse and human HD brain involving histone H3, trimethylation at lysine 4 (H3K4me3) and the potential efficacy in restoring epigenetic balance by targeting the relevant chromatin modifying enzymes. We find similar epigenetic patterns in differentiated HD iPS cells, establishing a “disease signature” that may guide transcriptional dysregulation in HD. The overall aims of this project are to; 1) generate data for cell signatures from human iPSC derived neurons, astrocytes and oligodendrocytes from healthy and diseased patients at baseline and in response to perturbagens with transcriptomics, epigenomics, whole-genome sequencing, proteomics and cell-based assays, including high-content longitudinal single-cell analysis. 2) Build cell signatures that convey the key features that distinguish the state of a cell and determine its behavior.Build simple signatures from the analysis of datasets generated with each technology individually, focused on features that distinguish cell state and that can be generalized within the larger LINCS Consortium. Integrate simple signatures into more complex meta-signatures with predictive value to accurately predict cellular responses to perturbagens. 3) Integrate the resources and results generated at this LINCS site with the broader LINCS consortium. Develop robust data provenance structures, including a database with web-based searchable access. Researchers (Thompson Lab): R. Lim, M. Casale, J. Stocksdale, J. Wu.
Neuroregulatory Mechanisms of PIAS1 and Implications for Huntington’s Disease
(Collaborators: B. Davidson, J. Van Eyk, K. Shuai, P. Sarkar)
Protein modifications of the mutated protein (mutant Huntingtin) that causes HD may alter progression of disease and understanding the mechanisms involved are crucial to the development of effective therapeutic intervention. We are using an innovative approach to understand the relationship between a specific Huntingtin modification system, involving the E3 SUMO ligase PIAS1, and HD pathology. We are examining the biochemical, neuropathological, and behavioral alterations upon PIAS1 modulation in the HD mouse model R6/2 and performing longitudinal studies in presymptomic and symptomatic zQ175 knockin mouse models. In addition we are studying the behavior of the PIAS1 network in the context of mHTT to understand its role in HD pathology. Studies in mouse primary cortical and striatal neurons, microglia as well as human HD-derived induced pluripotent stem cells will assess the impact of acute knock down and overexpression of PIAS1 in the context of mHTT expression on protein accumulation, post-translational modification, neuroinflammation, protein clearance networks, and toxicity. Given the broad range of potential pathways impacted by PIAS1, we will use transcriptomic and other biochemical approaches following PIAS1 modulation in expanded repeat HTT-expressing neuronal and microglial cells to identify the key pathways and mechanisms involved. We will also perform a complementary experiment to determine if HD molecular and behavioral phenotypes develop in PIAS1-/+ mice implanted with mHTT expressing virus to further validate PIAS1 as relevant to HD pathogenesis. The rationale is to have PIAS1 haploinsufficiency prior to mHTT expression, as some CAG repeat dependent changes may occur during development. Finally we will examine the functional significance of PIAS1 domains in disease modifying pathways. The PIAS protein family contains several highly conserved regions including the E3 SUMO ligase domain and an acidic region containing a putative SUMO1 and polySUMO interaction motif (SIM). We hypothesize that these domains may provide distinct contributions to disease phenotypes in HD systems and provide unique binding pockets for the rational design of PIAS1 inhibitory drugs. We will test this by mutating these motifs and assessing their contribution to disease phenotypes in primary neurons expressing mHTT and in vivo in R6/2 mice. Researchers (Thompson Lab): J Ochaba, C Geater.
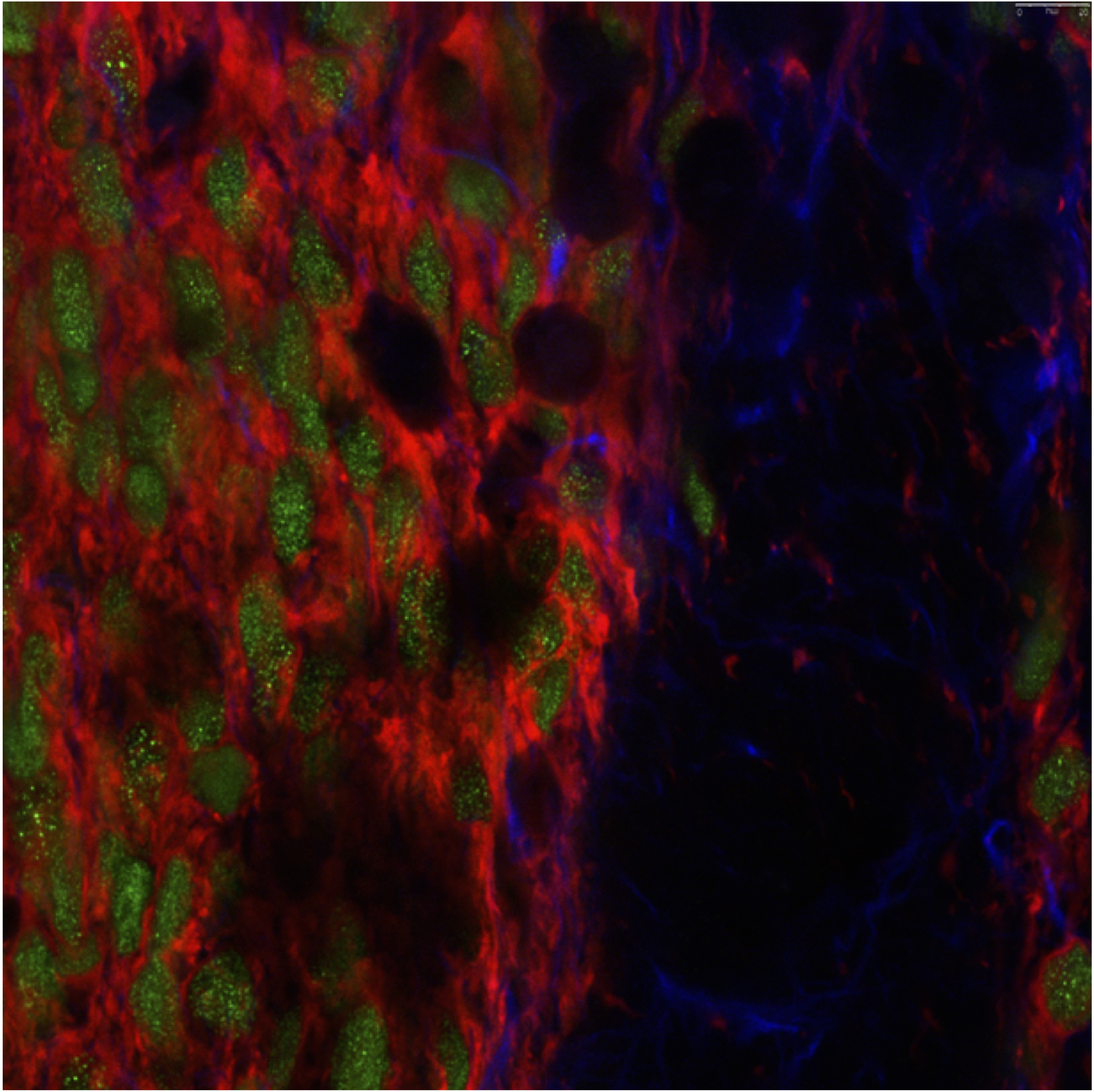
A hNSC Development Candidate for Huntington’s Disease
CIRM Clin 1 (Collaborators: M. Levine, M-F Chesselet, UC Davis (G. Bauer, B. Fury), AgeX, Biologics Consulting Group. Other: A. Morenkova, E. Monuki, Advisors: C. Svendsen, B. Leavitt, S. Tabrizi)
Human neural stem cells (green) implanted in a Huntington’s disease model mouse are co-stained with a marker for immature neurons (red).
Given the complexity of HD neuropathology and clinical impairment and the multiple systems affected, it seems unlikely that small molecule therapies only will sufficiently target each of the molecular and cellular processes affected during the unrelenting progression of this disease. We are testing cell-based approaches to provide a more effective method; delivering multiple therapeutic activities to several affected brain regions. We are producing and characterizing a scalable bank of human neural stem cells hNSCs for preclinical studies. Preclinical efficacy of the hNSCs in R6/2 HD mice will be performed including optimization of effective route of administration and dose range. In addition, we will perform long-term studies in pre symptomatic and symptomatic zQ175 knockin HD mice. Pharmacology and pilot safety studies will be also performed. The results of these studies are intended to be used in regulatory pre-IND meetings with the FDA for evaluation of hNSCs as a development candidate for clinical trials. Researchers: J. Reidling, S. Yeung, A. Lau, D. Sharifibad, P. Nguyen and A. King.