









The role of environmental risk factors in Alzheimer’s disease
Alzheimer disease (AD)
Alzheimer disease (AD) is a progressive neurodegenerative disorder that causes over 60% of all dementia cases in the elderly population. Currently, over 35 million individuals worldwide and over 5 million in the U.S. suffer from AD, and the number of patients is expected to increase dramatically over the next 40 years particularly in developed countries where the average longevity and ratio of aged population are becoming more prominent.
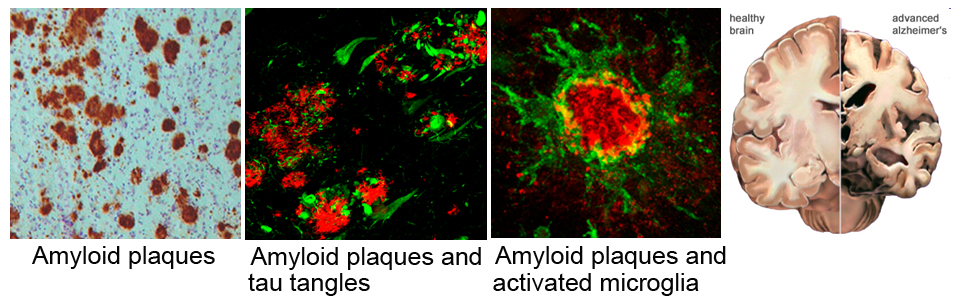
Clinical symptoms of AD include cognitive memory loss, particularly inability to recall recent events in the early stage of the disease, and gradual impairments in other domains that interfere with mood, reason, judgment, and language as the disease progresses. Eventually, even simple tasks such as maintaining personal hygiene cannot be performed, and the patient becomes socially dependent. Pathologically, AD develops two hallmark pathologies; amyloid plaques and neurofibrillary tangles. Amyloid plaques are extracellular deposits mainly composed of fibrillar forms of amyloid-beta (Aß), 39-43 amino acid peptides generated by the sequential proteolytic cleavage of amyloid precursor protein (APP), whereas neurofibrillary tangles are intraneuronal aggregates of hyperphosphorylated tau protein. Other neuropathological changes including inflammation, oxidative damages, mitochondrial dysfunction and profound neuronal cell death are also typically observed in the AD brain.
We have investigated the role of inflammation in AD pathology using transgenic mouse models of the disease. These mouse models develop amyloid plaques and/or tau tangles in an age-dependent manner.
Research Project 1: Gene x Environment interactions in the pathogenesis of AD
My laboratory is particularly interested in elucidating the pathogenic role of environmental risk factors that exhibit neurotoxicity leading to neurodegeneration and AD. We are currently investigating the following environmental contaminants:
1. Copper in drinking water – Although copper is an essential metal, excess or deficiency of copper causes neurological damage. It has been shown that chronic exposure to copper may increase the risk for AD. Elevated “free” copper in plasma correlates well with accelerated cortical thinning, cognitive decline and earlier onset of AD in humans. We are investigating how chronic low-dose copper exposure disturbs microglia function and vascular integrity in the brain using transgenic animal models.
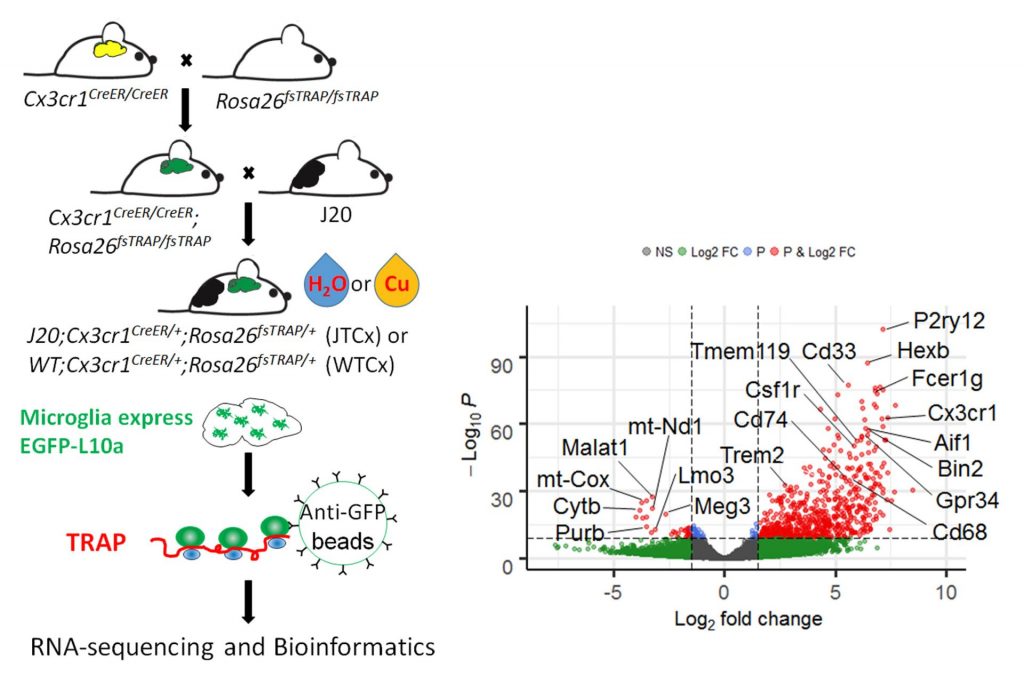
We use TRAP (translating RNA affinity purification) to isolate microglia mRNA from fresh mouse brain (Left). RNA-seq validates enrichment of microglia-specific mRNAs in the TRAP samples (Right).

Comparing TRAP samples between copper-exposed mice and control mice shows early polarization of microglia towards degenerative transcriptomic phenotypes.
2. Air pollution – How fine and ultrafine particulate matter (PM), volatile organic compounds (VOCs), metals, and polycyclic aromatic carbons (PAHs) alter brain inflammatory responses and oxidative stress, leading to degenerative process. In collaboration with Dr. Michael Kleinman’s lab at UCI, we expose mice to concentrated PM/VOCs/PAHs at various ages using VACES (below) and examine brain function and pathology.

Research Project 2: Evaluating drug candidates to treat AD in pre-clinical studies
Our laboratory is also interested in developing and evaluating drug candidates to treat AD symptoms using animal models. We have several on-going pre-clinical studies:
- Targeting inflammatory resolution to restore glial homeostasis in the brain: Our studies strongly suggest that dysregulation of brain inflammation triggers and/or exacerbates AD neuropathology. If we restore homeostasis of brain inflammation, we may ameliorate the progression of the disease process. In this regard, we identified the inflammatory resolution pathway as a potential pharmacological target to restore brain inflammation. Endogenous lipid mediators, such as lipoxins and resolvins, activate the resolution pathway. In collaboration with a pharmaceutical company, we are currently evaluating a novel compound that activate this pathway.
- Targeting monoamine neurotransmitters to restore cognitive functions: Recent studies unveil that extensive loss of tyrosine hydroxylase (TH)-positive neurons is evident in prodromal and early stage of AD. TH+ neurons in locus coeruleus (LC) project to the hippocampus and other brain regions and modulate synaptic plasticity, function and cognition. This leads to a hypothesis that loss of monoamine neurotransmitters input to the hippocampus causes cognitive decline and other symptoms in early stage of AD. We will be testing whether a drug to restore brain monoamine neurotransmitters effectively rescues or improve cognition in various animal models of AD.