The past 25 years of research have highlighted the production of the Aβ peptide as being the upstream cause of Alzheimer’s disease (AD), with its accumulation leading to a cascade of events ultimately resulting in synaptic loss followed by neuronal death. This brain atrophy underlies the dementia seen with the disease. Hence, for an individual to develop AD they must first accumulate Aβ within their brains, and then this Aβ must interact with the local brain environment to cause synapto- and neuro- toxicity. The disease selectively affects the aged brain and prevalence increases with increased age. Thus, the more youthful brain appears to be protected from the accumulation of the Aβ peptide, and its downstream effects, while the aged brain is more permissive of the accumulation of Aβ, and this permissiveness increases with further brain aging.
The crucial question is what changes in the brain as it ages to allow for the accumulation of Aβ peptide, and for its downstream toxic effects to be enacted? Over the past 20 years aberrant calcium dysregulation has been consistently implicated in AD, either in the initiation of the disease, the progression of AD-related pathologies, or in mediating the neuronal and synaptic loss that results from the presence of the pathologies.
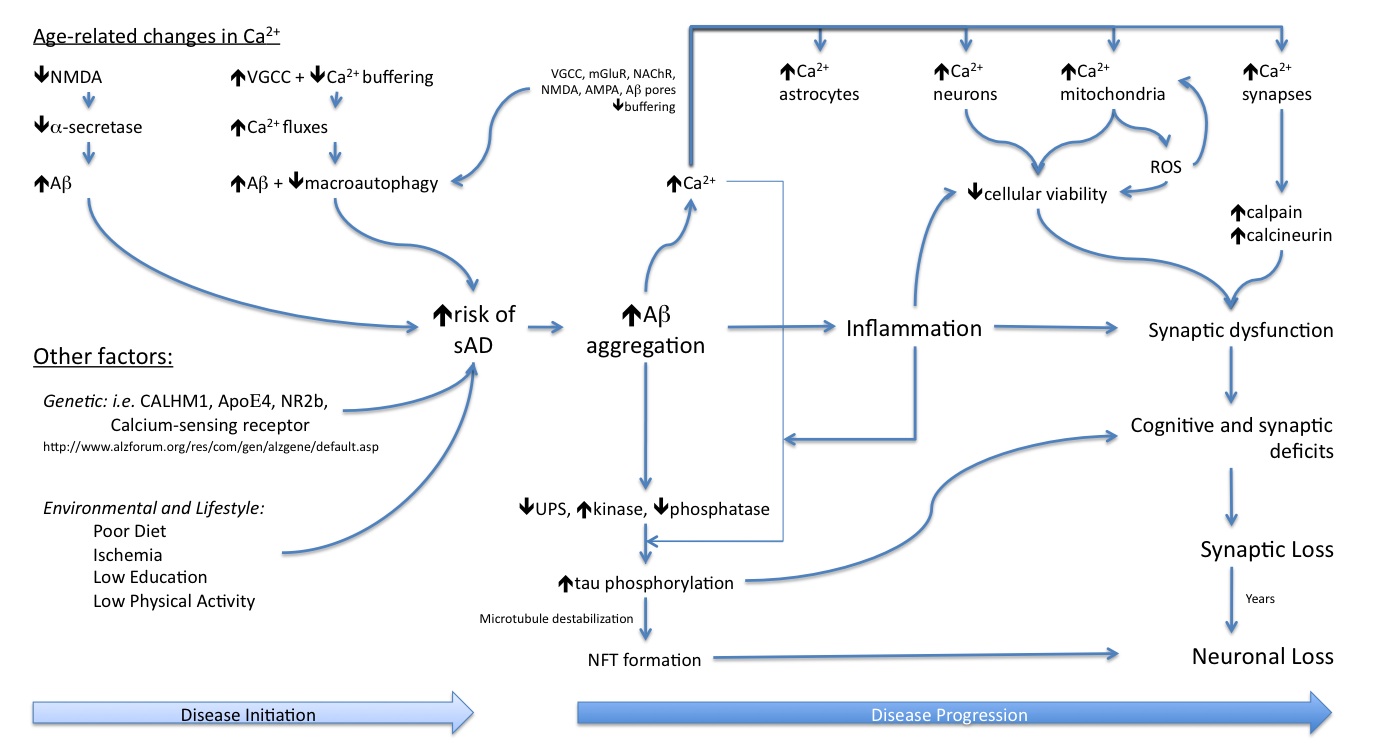
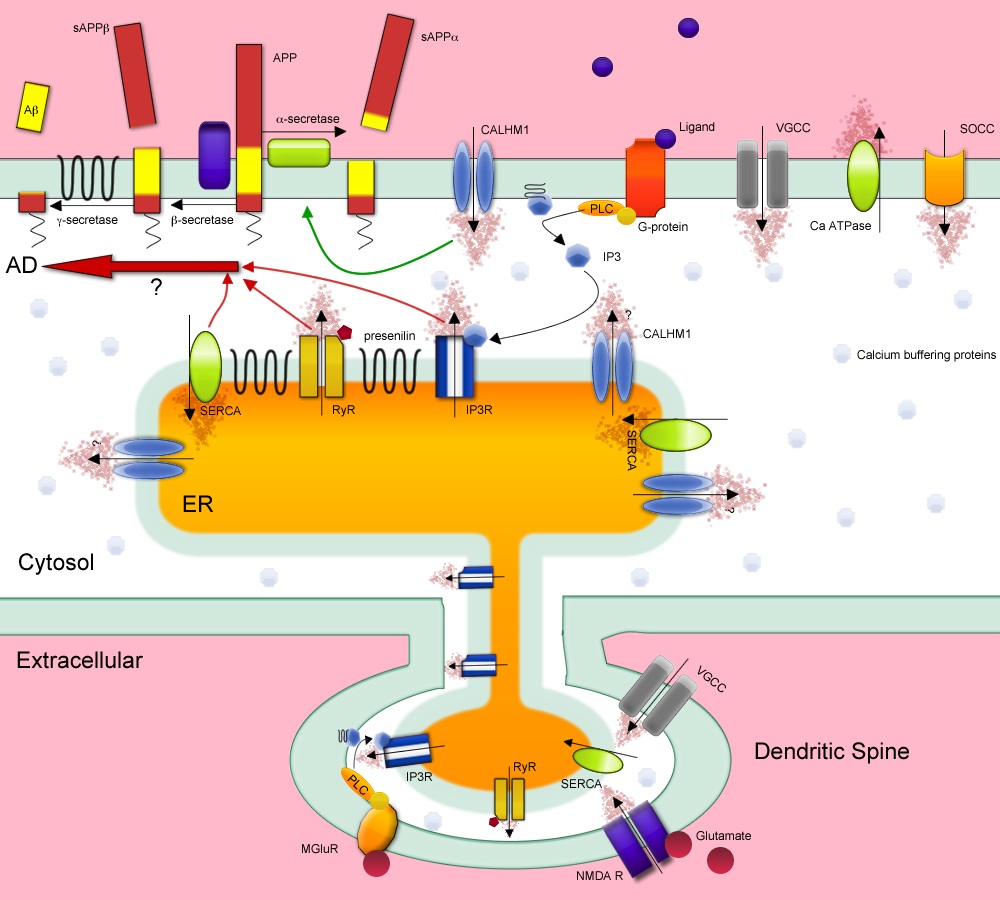
We have been investigating how several specific calcium sources can modulate APP processing and influence the development of AD pathology in the aged brain.
T-type Voltage-Gated Calcium Channels:
There are three types of T-type voltage gated calcium channels, designated as CaV3.1, CaV3.2, and CaV3.3, encoded by the genes cacna1g, cacna1h, and cacna1i, respectively. One or more types are expressed in neurons in most brain regions, including the hippocampus. In addition to neurons, T-type channels are found in the peripheral nervous system, the heart, smooth vascular muscle, and in various endocrine tissues. In the brain, T-type channels regulate neuronal firing patterns. They open in response to very negative membrane potentials, and are thus suited for regulating excitability. Furthermore, it means that even small graded depolarizations can open T-type channels causing rapid influx of calcium into the neuron, which itself can cause further depolarization and lead to an action potential, and also act as a second messenger signaling molecule. The channels inactivate rapidly, thus resulting in a transient influx of calcium with each depolarization from baseline, but can be reactivated with hyperpolarization.
T-type channels are located throughout the soma and dendrites of neurons, and thus provide a neuron-wide source of calcium influx. Because T-type channels can open with very small depolarizations, or even at resting membrane potential, they are a source of basal calcium influx into the neuron.
Regulation of neuronal firing patterns by T-type channels is crucial to normal physiology, in particular during sleep rhythms, and movement, as well as normal firing patterns in a number of neuronal circuits. Notably, T-type channels have been implicated in a number of disease states including epilepsy, autism, pain, and psychoses, as well as hypertension and heart failure in the periphery. As such T-type channels play a crucial role in the regulation of many systems in the CNS, and their dysfunction is implicated in a number of diseases. As our preliminary data show a robust reduction in T-type channel expression in the aged brain, it suggests that many of these systems may be impacted during aging.
Activating T-type channels as a therapeutic for Alzheimer’s disease:
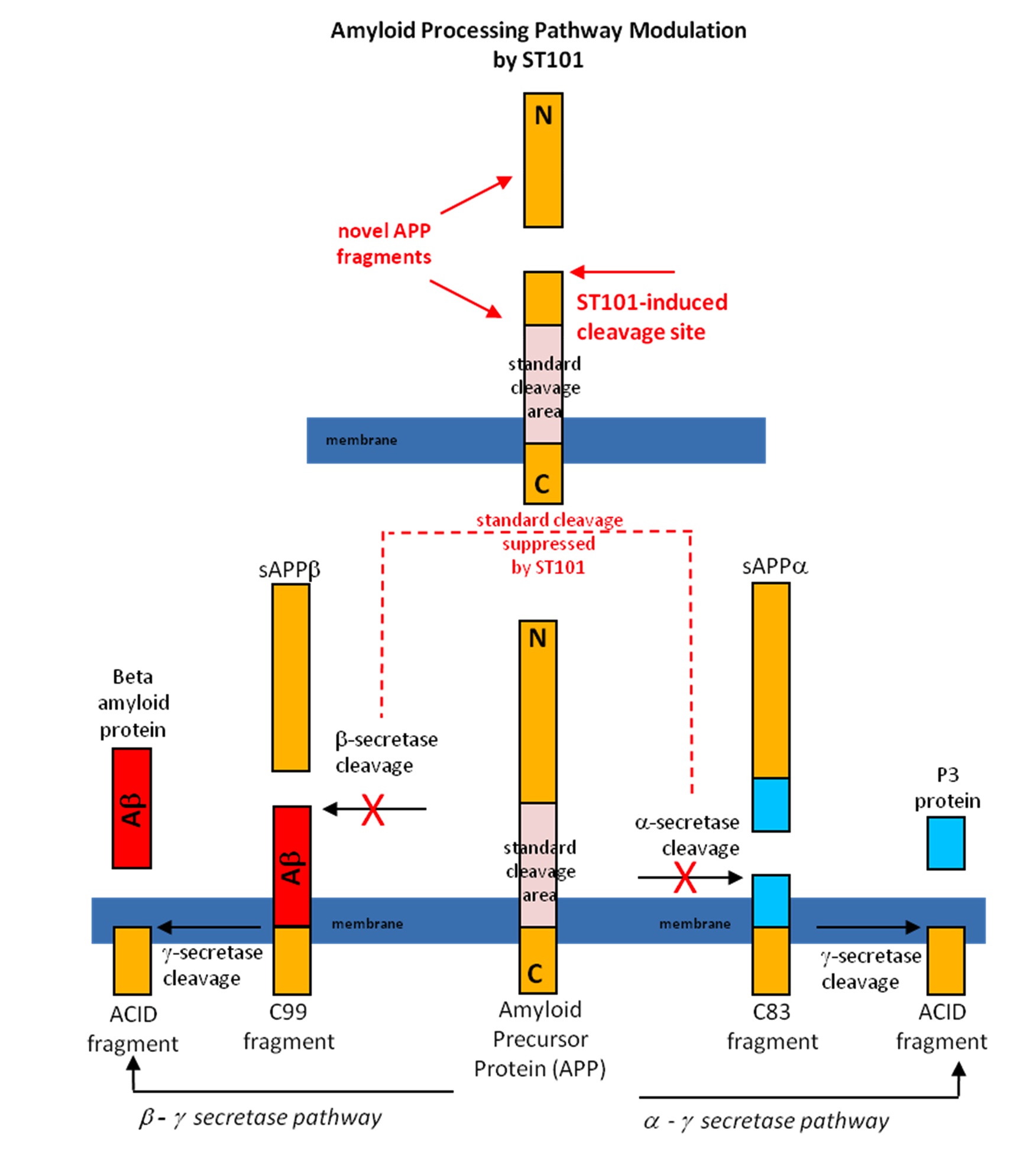
ST101 is a small molecule that acts as a T-type calcium channel activator. We found that that ST101 induces cleavage of APP at a novel site, generating a 17-kDa C-terminal fragment. This 17-kDa APP cleavage product does not appear to be a substrate for either a- or b-secretase, and thus bypasses generation of Ab. ST101 is orally active, efficacious at low doses, improves memory function and we found it robustly reduces brain Ab in transgenic mice and also non-human primates.

The following is a schematic of how the T-type channel activator ST101 modulates APP processing and bypasses Ab generation in the brain.
Two-Pore Channels and Alzheimer’s Disease:
We have recently shown that presenilins are crucial for efficient proteolysis via autophagic processes. Autophagy is the process by which cellular constituents like protein aggregates and damaged organelles are brought to the lysosome for degradation. Proper autophagy is vital to cell survival, neuronal health, clearance of microbes, long-lived proteins, and protein aggregates. Given this, autophagy is being actively pursued as a target for maintaining proper cellular health, especially to facilitate the clearance of protein aggregates. Our previous data indicated that presenilins function in autophagy through lysosome fusion to autophagosomes or lysosome function. Of note, calcium fluxes have been shown to regulate autophagy induction, especially via the IP3 receptor.
In addition to calcium fluxes regulating the induction of autophagy, lysosomes themselves act as calcium stores, and possess energy dependent calcium efflux channels, known as two-pore channels (TPC). TPCs are sensitive to the second messenger nicotinic acid adenine dinucleotide phosphate (NAADP). The two forms of TPCs in mammals are TPC1, which localizes to both endosomes and lysosomes, and TPC2, which appears to localize to lysosomes only. These channels are predicted to have two repeated domains of 6 transmembrane regions each, a structure that corresponds to half a voltage sensitive Ca2+/Na+ channel. Due to this, it is speculated that TPCs function as dimers. Additionally, TPC function seems to be negatively regulated by N-glycosylation near their pore sites. Little is known about the role of these channels in lysosome function, or on the autophagic processes that deliver autophagosomes to the lysosome. Fusion of organelles within the endo-lysosomal system is calcium dependent and therefore lysosomal calcium stores may play a role in autophagy.
We have been investigating the role of TPCs in both presenilin dependent regulation of autophagy, as well as their role in APP processing.